Cone rod dystrophy
- 19 Şub
- 3 dakikada okunur
Güncelleme tarihi: 9 Nis

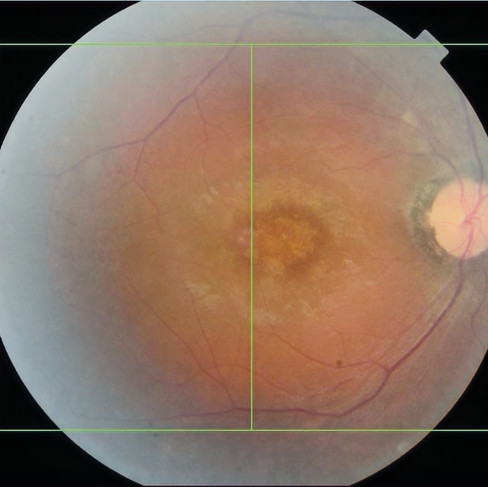


22 yaşında bayan hasta, görme her iki gözde de 0.3 seviyesinde, giderek görmesinin azaldığını ifade ediyor. Her iki gözde de periferik retinada yaygın bone-specule pigment birikimleri var. Retina arterleri belirgin daralmış ve optik diskler soluk olarak izleniyor. Bu klinik tabloyu tipik Retinitis Pigmentosa dan ayıran bulgu ise hastanın her iki makülasında belirgin retina pigment epitel değişikliklerinin olması... Makülada retina pigment epitel atrofisine bağlı depigmente alanlar dikkati çekiyor. Makülada pigment epitel atrofisinin yanı sıra fovea etrafında refle artışı olması hastanın yaşının çok genç olması nedeni ile vitreus kaynaklı bir refle artışıdır ilerleyen dönemlerde bu kaybolacak ve yerini burada da görülecek olan retina pigment atrofisine bırakacak.


Her iki gözün OCT incelemesinde de neredeyse birbirinin aynı bulgular dikkati çekiyor. Her iki gözde de tüm kesitlerde ISOS bandı ve ELM tamamen kaybolmuş. Retina incelmiş, retina pigment epiteli yer yer kabalaşmış, yer yer atrofik alanlar dikkati çekiyor. Bunlar tipik retinitis pigmentosa bulgusu fakat bu hastaya özgü olan bulgu ise bu bulguların foveada da izlenmesi, yani maküla ve periferik retinada hemen hiç bir alanda fotoreseptör kalmamış.
İleri evre Retinitis Pigmentosa veya Rod cone distrofilerde de hastalığın son evrelerinde foveada da atrofi gelişmesini sıklıkla görürüz. Normalde Retinitis pigmentosa' da periferik retinadaki rod hücrelerinin %70 inden fazlası atrofiye uğradığında santraldeki kon hücrelerinin de apoptozise gitmeye başladığını görüyoruz. Bu durumda bu hastanın ileri evre Retinitis Pigmentosa dan ayırıcı tanısını nasıl yapacağız?
Her ne kadar bu hastanın yaşının 22 olması ve Retinitis Pigmentosa'nın son evresine geçmiş olduğunu düşünmemiz için çok erken bir yaşta olduğunu göz önünde bulundurmamız gerekirse de iyi bir ayırıcı tanı yapabilmek için önce odaklanmamız gereken unsur hastanın yakınmalarıdır. Bu hasta gece körlüğünden değil, çocukluğundan beri gündüz bol ışıkta daha az gördüğünden yakınıyor. Bu bulgu bize kon hücre baskın bir hastalıkla karşı karşıya olduğumuzu düşündürüyor.
İkinci unsur en azından biz hastayı gördüğümüzde OCT incelemesinde rod ve kon hücrelerinin eşit durumda etkilenmiş olduğunu tespit etmemizdir.
Hastalığın bu evresinde ERG araştırmalarının bize iki hastalığı ayırt etmekte yararı çok az olacaktır. Bu hastaya ERG tetkiki yapıldı ve hem karanlık adaptasyonda hem de fotopik ve flicker yanıtlarında hemen hemen hiç elektriksel aktivite ve dalga oluşumu tespit edilmedi. Hastayı daha erken evrede görseydik ERG bulguları daha çok yol gösterici olabilirdi.
Genetik testler teorik olarak bize ayırımda yardımcı olabilir fakat günlük pratiğimizde bunun yararı da çok az oluyor;
Genetik Spektrum cone ve cone rod dystrophy
Hastalığın fenotipi, altta yatan mutasyona göre dramatik şekilde değişir.
ABCA4: En sık görülen mutasyondur. Stargardt hastalığı ile fenotipik örtüşme gösterir (KRD fenotipi baskındır).
GUCY2D: Otozomal dominant KD vakalarının çoğundan sorumludur. Erken yaşta ciddi fotopik ERG kaybı ile karakterizedir.
CRX: Genellikle şiddetli seyreden, erken başlangıçlı fotoreseptör dejenerasyonları ile ilişkilidir.
PROM1: Hem KD hem de maküler distrofilere neden olabilen yapısal bir protein mutasyonudur.
Bu klinik görünümü bir de kon distrofiden ayırt etmemiz gerekir. Kon distrofilerde de izlenen hasar fovea ile sınırlı kalmaz ve makülanın tamamına ve perifere doğru ilerleme eğilimindedir. Geç olgularda bunları ayırt etmek iyice güçleşir, bunun ayırıcı tanısı ise şöyle bir tabloda özetlenebilir.
Ayırıcı Tanı
Parametre | Kon Distrofisi (KD) | Kon-Rod Distrofisi (KRD) |
İlk Semptom | Fotofobi ve santral görme kaybı | Fotofobi ve renk görme bozukluğu |
Gece Körlüğü | Genellikle görülmez veya çok geç evre | Erken/Orta evrede görülür |
Periferik Görme | Uzun süre korunur | Erken dönemde daralmaya başlar |
Fundus | Bull's Eye makülopati baskındır | Makülopatiye ek olarak kemik spikülleri (periferde) |
ERG | İzole fotopik kayıp, skotopik normal | Hem fotopik hem skotopik kayıp (fotopik > skotopik) |
Klinik pratikte temel fark, rod (çubuk) hücre tutulumunun varlığı ve zamanlamasıdır. KD'de fonksiyon bozukluğu kon hücrelerine sınırlıyken, KRD'de başlangıçta konlar etkilenir ancak bunu kaçınılmaz bir rod dejenerasyonu izler.
Sonuç olarak ayırıcı tanı yapılması zor olan bu hasta grubunda klinik bulgular ve tetkiklerin yanı sıra hastaların yakınmalarının da değerlendirilmeye katılması çok önemlidir.
Kaynakça
Gill, J. S., et al. (2019). "Inherited Cone and Cone-Rod Dystrophies: Phenotype-Genotype Correlations." Surv Ophthalmol.
Zelinger, L., et al. (2015). "Identification of a Novel Mutation in ABCA4 Associated with a Rare Form of Cone-Rod Dystrophy." Investigative Ophthalmology & Visual Science (IOVS).
Kohl, S., et al. (2012). "Genetics of cone and cone-rod dystrophies." Progress in Retinal and Eye Research.



Yorumlar